Prediction
Prediction.RmdOverview
Perhaps the most obvious use after we have created models like GWAS, GWAX and LT-FH would be to see if we were able to predict whether a new person, with a genotype that has never before been seen, would be a case. There are many ways to predict, and this package in no way contains an exhaustive method of prediction, but we do take some steps to quality control our predictions. The simplest method of predicting would no doubt be using GWAS on the entire dataset, then matrix multiplying the resulting vector of effect sizes on a new person’s SNPs, resulting in a single number, their Polygenic Risk Score (PRS), which could then be converted to a status. The resulting model for predicting would probably not be very good, maybe a little better than randomly guessing. What steps can we take to improve our predictive model? For starters, we are currently using all SNPs no matter how insignificant they are (how high their p-values are in the regression). We aren’t sure which significance level is best to threshold at, so we can create a vector of all the alpha levels we wish to test our thresholding at. Now we can get a model for predicting for every single one of these thresholds, each one including a different amount of SNPs. That solves the problem of how many SNPs to include, but there are other issues to solve.
Cross-validation
We want to avoid overfitting, but we also want to avoid having to simulate lots of new data to test for overfitting. That’s why we make use of k-fold cross-validation. We split the data into k equal parts, train our predictive model on k-1 parts, and then test the model on the remaining part of the data. Then we repeat for the remaining parts, until we have trained and tested k times. We can then choose the best model for predicting, and if we have done sufficient cross-validation, we can assume that it is not overfitted to the training data. You can in theory choose k to be any number between 1 and n, with n being the number of genotypes. A k = 1 would mean no cross-validating at all, and k = n would mean that the model is trained on n - 1 people and tested on 1 person, n times. Our implementation of cross-validation does not allow for k = n, as the scoring system cannot work with too few data points at a time. k = 10 should be sufficient in reducing overfitting in most cases, and as long as the dataset isn’t too small.
Choosing the Best Model
We can now see what would be the best alpha value with simple GWAS, but what’s stopping us from utilizing our GWAX proxy statuses or our LT-FH posterior mean liabilities? Nothing! We simply change what we are regressing on, but our methodology doesn’t change otherwise.
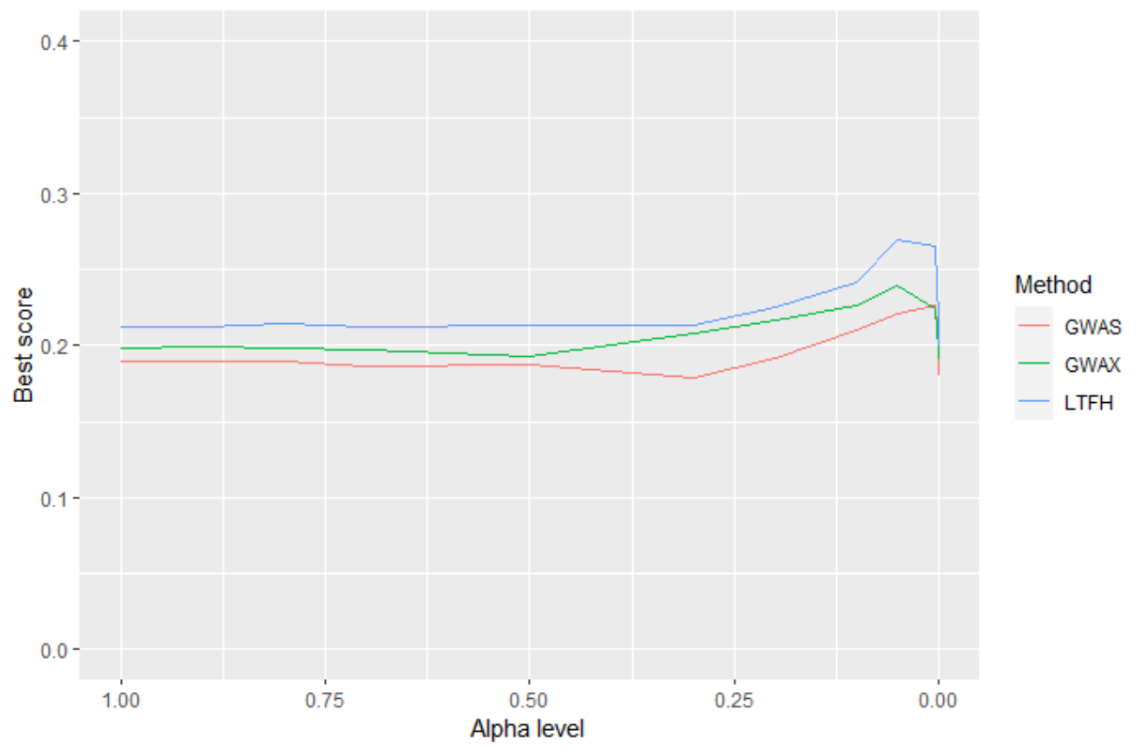
We can easily compare and choose the best model for predicting as seen in the above figure, where we illustrate the results from Prediction_cross_validation() on the three methods, where a higher score means better prediction. Note that the alpha level is decreasing on the x-axis. We can now predict a status for a new person and be comfortable that we have the best model that we can create to do so.
Though we output statuses, we are actually calculating PRS, which means we can do even more behind the scenes, like trying to minimize false positives by only assigning someone to be a case if they have a very high PRS. When using LT-FH, we can even attempt to guess how many cases and non-case family members a person has, though we should be careful if our model is not already very robust, as the number of possible family combinations also increases rapidly with more siblings, and some configurations can have similar posterior mean genetic liabilities.